






It’s been five years since COVID-19 was declared a global pandemic. As SARS-CoV-2 shifts to endemic status, questions about its future evolution remain. New variants of the virus will likely emerge, driven by positive selection for traits such as increased transmissibility, longer infection duration and the ability to evade immune defenses. These changes could allow the virus to spread among previously immunized populations, potentially triggering new waves of infection.
Predicting new mutations in viruses is crucial for advancing life science research, particularly when trying to understand how viruses evolve, spread and affect public health. Traditionally, researchers rely on wet-lab experiments to study mutations. However, these experiments can be costly and time-consuming.
Researchers from the College of Engineering and Computer Science at Florida Atlantic University have developed a new method to predict mutations in protein sequences called Deep Novel Mutation Search (DNMS), a type of artificial intelligence model that uses deep neural networks.
For the study, they focused on the SARS-CoV-2 spike protein – the part of the virus responsible for helping it enter human cells – and used a protein language model to predict potential new mutations in this protein never seen before.
To do this, researchers used a language model, ProtBERT, which was specifically fine-tuned to understand the “dialect” of SARS-CoV-2 spike proteins. The model works by looking at potential mutations and ranking them based on several factors. These include grammaticality, which refers to how likely or “correct” a mutation is according to the grammatical rules learned by the model, as well as how similar the mutated sequence is to the original protein, which is measured by semantic change and attention change.
Results of the study, published in the journal Communications Biology, show that the DNMS language model can separate sequences into groups based on their similarities. The model can predict which mutations are likely to occur by looking for mutations that cause only small changes in the protein’s structure and function. This is important because, in most cases, viruses like SARS-CoV-2 evolve through small changes that allow them to adapt without drastically altering their overall function.
The DNMS method uses all available information about the sequence and the mutations to create a more accurate prediction of which mutations are likely to occur. Unlike prior research, which typically looks at changes to a reference protein sequence, DNMS introduces a parent-child mutation prediction model. The parent sequence (an existing protein sequence) is used to predict mutations, and these mutations are analyzed based on how they might evolve over time.
“Our model ranks all possible mutations to find the ones that are most likely to occur in the future,” said Xingquan “Hill” Zhu, Ph.D., senior author and a professor in FAU’s Department of Electrical Engineering and Computer Science. “Our study shows that mutations following the protein’s grammars, with minimal changes compared to the original sequence and low attention differences, are considered the most likely future mutations.”
The method first takes a given SARS-CoV-2 spike protein sequence and simulates all possible single-point mutations. For each mutated version of the protein, DNMS uses the ProtBERT model to calculate how likely each mutation is to follow the “grammar” of the protein (grammaticality) and how similar the mutated sequence is to the original sequence (semantic change). Additionally, the model looks at attention, a measure that has been used to study protein structure and function, but never before applied to mutation prediction.
“The key to our method lies in using the context provided by the parent sequence. This context is crucial for evaluating whether a potential mutation aligns with the ‘grammar’ of the protein,” said Zhu. “DNMS works by selecting a parent sequence from a phylogenetic tree – basically a family tree of viral strains – and simulating all possible mutations.”
The study also looked at the relationship between the predicted mutations and the virus’ fitness, or how well it can replicate and survive. Findings show that mutations with high grammaticality, small semantic change, and low attention change were associated with higher viral fitness. This suggests that mutations which fit well within the biological “rules” of the protein and cause minimal disruption to the protein’s structure or function are more likely to be beneficial for the virus.
“We believe that using sequence data alone can help make these predictions, as proteins follow certain biological rules,” said Zhu.
The researchers tested the effectiveness of DNMS through statistical analysis. Their results show that DNMS outperforms other methods in predicting novel mutations because it combines all the relevant factors into a single, more accurate prediction model.
“The fine-tuned, pre-trained language model developed by our researchers can predict which SARS-CoV-2 mutations are more likely to occur in the future,” said Stella Batalama, Ph.D., dean of the College of Engineering and Computer Science. “This method can be useful for guiding experimental research, as it provides predictions about mutations before they are observed in the population, helping public health officials track and prepare for new mutations before they spread widely.”
Study co-author is Magdalyn E. Elkin, a doctoral student in FAU’s Department of Electrical Engineering and Computer Science.
The research was sponsored by the United States National Science Foundation.
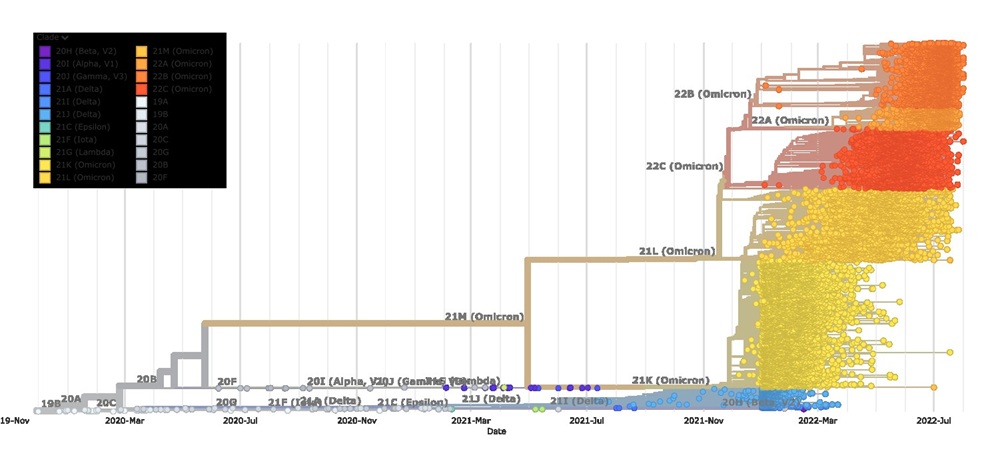 A phylogenetic tree of SARS-CoV-2 sequences, built with Nextstrain, displays sequences colored by their clade and organized by collection date. Over time, sequences mutate and diverge, forming new clades, much like dialects in natural languages. Unlike traditional methods that focus on mutations from the original reference sequence, this new research incorporates a parent-child relationship in virus evolution. It evaluates mutations not only in relation to the reference sequence but also by assessing their impact on the grammaticality, semantics, and attention of the protein sequence to identify the most significant mutations.
A phylogenetic tree of SARS-CoV-2 sequences, built with Nextstrain, displays sequences colored by their clade and organized by collection date. Over time, sequences mutate and diverge, forming new clades, much like dialects in natural languages. Unlike traditional methods that focus on mutations from the original reference sequence, this new research incorporates a parent-child relationship in virus evolution. It evaluates mutations not only in relation to the reference sequence but also by assessing their impact on the grammaticality, semantics, and attention of the protein sequence to identify the most significant mutations.
-FAU-
Latest News Desk
- FAU Celebrates Spring 2025 GraduatesFlorida Atlantic University conferred more than 3,700 degrees over the course of seven in-person commencement ceremonies in the Carole and Barry Kaye Performing Arts Auditorium.
- FAU Joins First Global Effort to Map Microplastics in Ocean SystemsAn FAU researcher joins an international team of scientists who have moved beyond "scratching the ocean's surface," marking a turning point in understanding microplastics' path through critical ocean systems.
- FAU Earns Military Friendly® Status for 2025-26Florida Atlantic University has been recognized as a Military Friendly® School, earning a silver designation among large public universities.
- FAU Student Wins Statewide Governor's Cup CompetitionA FAU engineering student recently won first place for his artificial intelligence code at the Roundtable of Entrepreneurship Educators of Florida's (REEF) 2025 Governor's Cup Entrepreneurship Competition.
- FAU CA-AI Lands $2.1M to Form New U.S. Air Force Center of ExcellenceTo address critical U.S. Air Force communications needs, FAU engineering's CA-AI has received a $2.1 million grant from the U.S. Department of Defense Air Force Research Laboratory.
- Nursing 2025: No Relief as Burnout, Stress and Staffing Woes PersistA new national survey from Cross Country and FAU of 2,600 nurses and nursing students paints a sobering picture of a profession at a breaking point and an urgent call to action.